How SaMD is Regulated in the US
There are many different medical markets around the world, with the largest two being the US and the EU. In this article, we are going to focus on the regulation in the US.
SaMD is considered an actual medical device, so it is regulated in the US by the FDA just as traditional medical devices are. The first thing the FDA requires is a Quality Management System, or QMS. This is because for software to be considered a medical device, it must follow the Quality System Regulations (QSR) published by the FDA.
History of Regulation for SaMD
The first guidance on premarket submissions was actually published back in 2005. Since then, a new rendition of this guide was published in 2021. Unfortunately, this can make things a bit tricky because both the current guidance and the draft guidance list the documentation you must submit depending on the intended use of your SaMD.
The Differences in Guidelines for SaMD
The guidance from 2005 divides SaMD into 3 separate “levels of concern” categories. These levels of concern are based on how severe the device could injure patients if the device fails, is flawed, or is misused. The 3 levels of concern are:
- Minor - As the lowest level of concern, no injury will occur to the patient or the operator of the device if it stops working as it was designed to.
- Moderate - This level includes all devices that could cause minor injuries if the device is flawed or misused. The moderate level also incorporates minor injuries that occur due to misinformation from the device over time.
- Major - This is the most dangerous level of concern where the device could cause serious injury or even death if the device is flawed. Like the moderate level, it also includes injuries from misinformation or misuse over time.
These levels of concern are not to be confused with the device’s risk classification. To learn more about SaMD risk classification, read this post.
The 2021 guidance replaces the 3 levels of concern with 2 levels of documentation: Basic Documentation and Enhanced Documentation. Since there is no definitive indication of which is primarily used for approval, it’s a good idea to have both, at least until the guide has been finalized. This shouldn’t be much, if any, extra work, as the documentation you need for one will likely be very similar to the other.
How is Regulation Different in the EU?
Regulation in the EU doesn’t differentiate from the US guidance regarding the software being treated as a traditional medical device. One noticeable difference, however, is what the software is referred to as. In the US, we know this type of software is called Software as a Medical Device (SaMD), but in the EU this software is referred to as Medical Device Software (MDSW).
Since this article is about regulation in the US, I won’t get into the specific requirements. There is other, different documentation that is needed to be considered MDSW in the EU.
Other Regulation Requirements
There are other requirements from the FDA which we have discussed in length in other blog posts. The first is risk classification, in which there are 4 categories:
- Class I
- Class II
- Class III
- Class IV
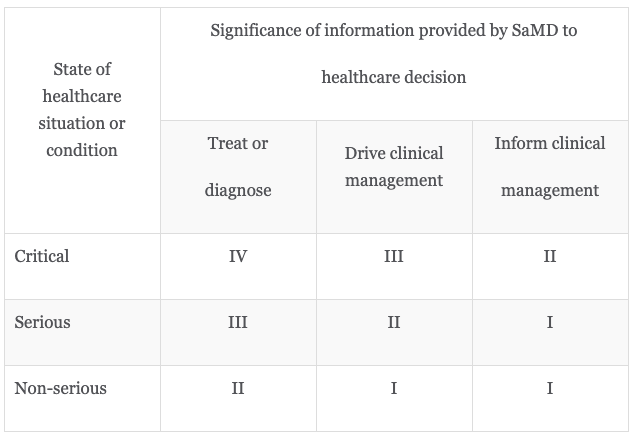
These classes are created from 2 identities including 1. the seriousness of the situation or health condition of the patient and 2. the significance of the information provided by the SaMD. Here is a chart for reference.
The last piece of regulation in the US has to do with the Software Safety Classification. It's important to understand that the software safety classification does not determine how safe your software is. Instead, it is the basis for determining how rigorous your software development process must be.
There are also 3 categories for the software safety classification of SaMD, which is based upon how serious of an injury would occur if the software failed.
- Class A - no injury
- Class B - non-serious injury
- Class C - death of serious injury
Classifying SaMD and getting approval from the FDA is a complex process which takes time to complete. These are just the major requirements, too! There are other requirements based on the different classes the device falls into.
If you are looking for assistance in planning a SaMD project, click the link below. We’ve been through the process before and we’ll help you come up with a strategic plan.
